We present first-principles density-functional calculation for the electric properties of boron (B)-, carbon (C)-, nitrogen (N)-, fluorine (F)-, phosphorus (P)-, and sulfur (S)-doped SrTiO3. The obtained results indicate that the bands originating from B (C, N, F) 2p or P 3p states appear in the band gap of SrTiO3, but the mixing of B (C, N, F) 2p or P 3p states with O 2p states is too weak to produce a significant band gap narrowing. Only in S-doped SrTiO3 case, the S 3p states mix well with the O 2p states and increase the width of valence-band (VB) of SrTiO3 which can produce the really band gap narrowing. Our results fully explain the absorption of visible light is due to the B (C, N, F) 2p or P 3p isolate states above the VB maximum of SrTiO3, while for S-doped SrTiO3 the visible light absorbance originating from the mixing of S 3p states with O 2p states which causing the band gap narrowing. We also propose that N (S)-doped SriO3 would be the best choice for single anion doping SrTiO3, while the B (C, P) elements may be the better candidates for co-doping.
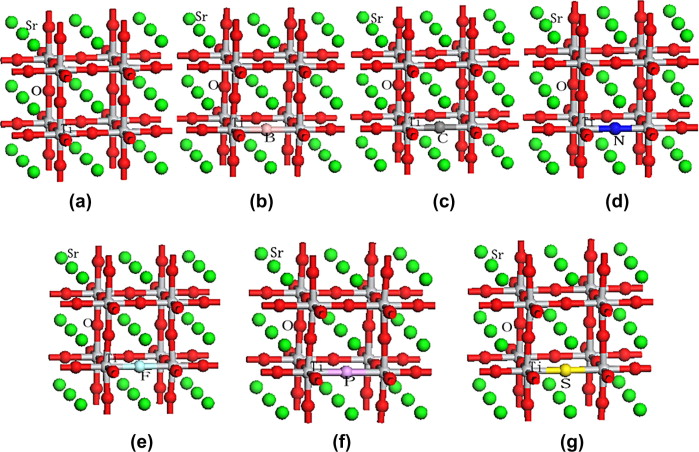
SrTiO3 super-cell models (a) un-doped SrTiO3, (b) one B atom doped 2 × 2 × 2 SrTiO3 super-cell, (c) one C atom doped 2 × 2 × 2 SrTiO3 super-cell, (d) one N atom doped 2 × 2 × 2 SrTiO3 super-cell, (e) one F atom doped 2 × 2 × 2 SrTiO3 super-cell, (f) one P atom doped 2 × 2 × 2 SrTiO3 super-cell, (g) one S atom doped 2 × 2 × 2 SrTiO3 super-cell. (Photo from the original paper)